Biopython – 序列文件格式
Biopython的 Bio.SeqIO 模块提供了广泛的简单统一的接口来输入和输出所需的文件格式。这种文件格式只能将序列作为SeqRecord对象处理。指定文件格式时使用小写字符串。 Bio.AlignIO模块也支持相同的格式。文件格式列表如下:File Format Description abi It is sequencing trace format for Applied Biosystem, reads the Sanger capillary sequence including PHRED quality scores for base calls. Each file contains only one sequence so no point of indexing the file. abi-trim Same as the abi format but has a quality trimming with Mott’s algorithm. ace Reads the overlapping or contiguous sequences from the ACE assembly file. cif-atom Used to figure out the partial protein sequence by structure based on atomic coordinates cif-seqres Determines the complete protein sequence by reading a macromolecular Crystallographic Information File(also known as mmCIF) as defined by the _pdbx_poly_seq_scheme record. embl EMBL(Protein and DNA seq file format) flat file format, uses Bio.GenBank internally. fasta A generic sequence file format, each record starts with a line starting with > character followed by other sequence lines. fasta-2line Strict interpretation of FASTA file format by no line wrapping(i.e. using two line per record). fastq A FASTA variant with Sanger used to store PHRED sequence quality values with an ASCII of offset 33. fastq-sanger Alias for FASTA having consistency with BioPerl and EMBOSS fastq-solexa Original Solexa/Illumnia interpretation of FASTA format which is used to encode solexa quality scores with ASCII having offset of 64. fastq-illumina Solexa/Illumnia 1.3 to 1.7 version of FASTA format used to encode PHRED quality scores with ASCII of offset 64. gck Local format used by Gene Construction Kit. genbank GenBank or GenPept flat file format. gb Alias for genbank, having consistency with NCBI Entrez Utilities. ig IntelliGenetics file format, appears to be same as the MASE alignment format. imgt EMBL variant format from IMGT, where feature tables are intentionally allowed for longer feature types. nib UCSC nib file format for nucleotide uses 4-bits(1-nibble) to represent one nucleotide(2 nucleotides require i byte). nexus NEXUS multiple alignment format, also called PAUP format. pdb-seqres Reads PDB(Protein Data Bank) file and determines complete data sequence by header. pdb-atom Determines partial protein sequence by structure based on atomic coordinates section of file. phd These files are output of PHRED, used by PHRAP and CONSED for input. pir FASTA variant created by NBRF(National Biomedical Research Foundation) for Protein Information Resource database(PIR), presently art of UniProt. seqxml Simple XML file format. sff Standard Flow gram Format binary files created by Roche 454 and IonTorrent/IonProton sequencing machines. sff-trim Standard Flow gram Format (SFF) applies trimming to listed files. snapgene Local format used by SnapGene. swiss Plain text Swiss-Pro also called UniProt format. tab Simple two column tab separated sequence file, each line stores record identifier and sequence. qual FASTA variant having PHRED quality values from sequencing DNA. uniprot-xml UniProt XML format replacement of SwissProt plain text format. xdna DNA Strider’s and SerialCloner’s local format, used by Christian Marck.
下面的实现解释了如何解析两种最流行的序列文件格式,即FASTA和GenBank 。
法斯塔:
它是存储序列数据的最基本的文件格式。最初FASTA是在生物信息学早期进化过程中创建的用于蛋白质和 DNA 序列比对的软件包,主要用于搜索相似性。下面是解析FASTA文件格式的简单示例:
例子:
要获取使用的输入文件,请单击此处。
Python3
# Import libraries
from Bio.SeqIO import parse
# file path/location
file = open('is_orchid.fasta')
# Parsing the FASTA file
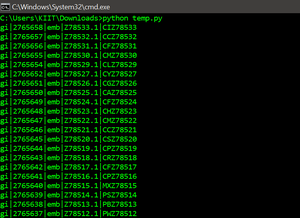
for record in parse(file, "fasta"):
print(record.id)Python3
# Import libraries
from Bio import SeqIO
from Bio.SeqIO import parse
# Parsing Sequence
seq_record = next(parse(open('is_orchid.gbk'), 'genbank'))
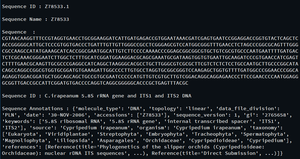
# Sequence ID
print("\nSequence ID :", seq_record.id)
# Sequence Name
print("\nSequence Name :", seq_record.name)
# Sequence
print("\nSequence :", seq_record.seq)
# Sequence Description
print("\nSequence ID :", seq_record.description)
# Sequence Annotations
print("\nSequence Annotations :", seq_record.annotations)输出:
基因库:
更丰富的基因序列格式,包括各种注释。解析GenBank格式就像更改Biopython解析方法中的格式选项一样简单。下面是解析GenBank文件格式的简单示例:
例子:
要获取使用的输入文件,请单击此处。
蟒蛇3
# Import libraries
from Bio import SeqIO
from Bio.SeqIO import parse
# Parsing Sequence
seq_record = next(parse(open('is_orchid.gbk'), 'genbank'))
# Sequence ID
print("\nSequence ID :", seq_record.id)
# Sequence Name
print("\nSequence Name :", seq_record.name)
# Sequence
print("\nSequence :", seq_record.seq)
# Sequence Description
print("\nSequence ID :", seq_record.description)
# Sequence Annotations
print("\nSequence Annotations :", seq_record.annotations)
输出: