Biopython – 序列比对
序列比对是将两个或多个 DNA、RNA 或蛋白质序列排列起来,以特异性识别它们之间的相似区域的过程。相似性的鉴定提供了很多关于物种间哪些特征是保守的、不同物种在遗传上有多接近、物种如何进化等信息。Biopython 具有广泛的序列比对功能。
读取序列比对: Biopython提供的Bio.AlignIo用于读取和写入序列比对。生物信息学中有很多可用的格式来指定序列比对数据与序列数据相似。 Bio.AlignIO有一个类似于Bio.SeqIO的 API,唯一的区别是Bio.SeqIO处理序列数据,而Bio.AlignIO处理序列数据比对。以下是下载示例序列比对文件的一些步骤:
- 首先打开浏览器,访问http://pfam.xfam.org/family/browse,可以按字母顺序查看所有Pfam家族。
- 现在选择任何具有较少种子值的家庭,因为它包含最少的数据并且易于工作。让我们用 PF18225 (http://pfam.xfam.org/family/PF18225) 移动一个。
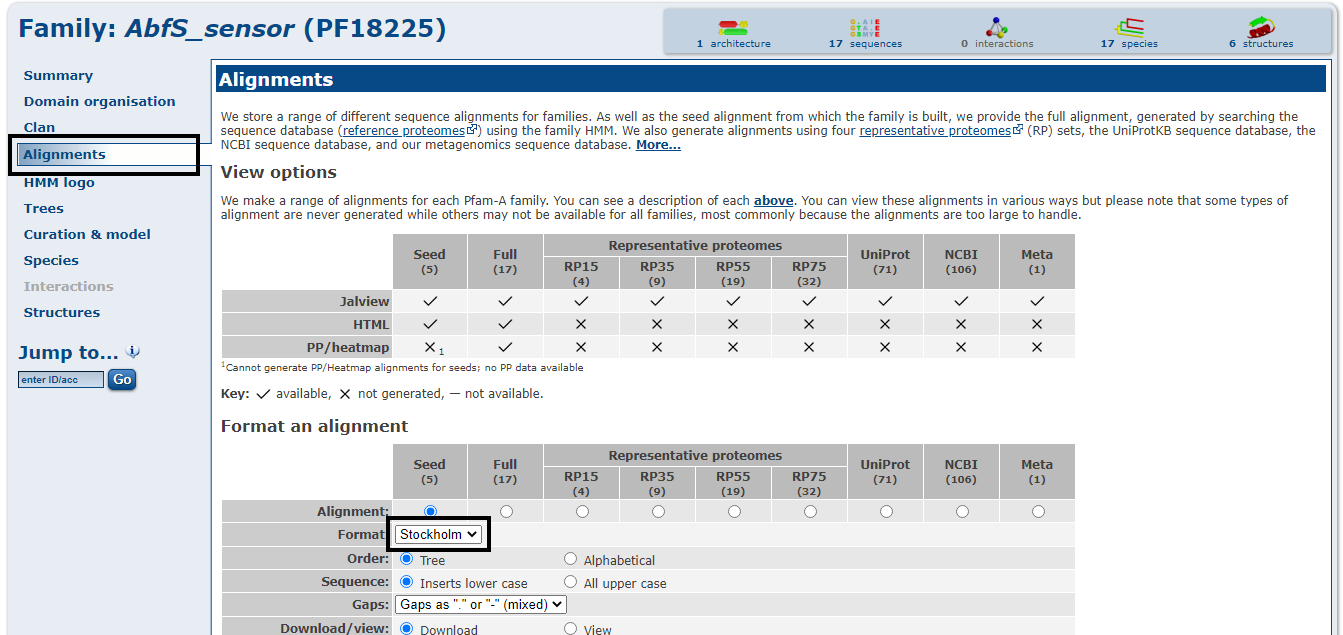
- 单击对齐部分并下载所需的斯德哥尔摩格式的序列对齐文件。
例子:
Python3
# Import libraries
from Bio import AlignIO
# Creating Sequence Alignment
alignment = AlignIO.read(open("PF18225_seed.txt"), "stockholm")
# Print alignment object
print(alignment)
# Show alignment sequence record
print("Showing Alignment Sequence Record")
for align in alignment:
print(align.seq)Python3
# Import libraries
from Bio import AlignIO
# Parsing Sequence Alignment
alignment = AlignIO.parse(open("PF18225_seed.txt"), "stockholm")
# Show alignment generator
print(alignment)
# Printing alignment
for alignment in alignments:
print(alignment)输出:
SingleLetterAlphabet() alignment with 5 rows and 65 columns
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM…NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA…NRT K4KEM7_SIMAS/61-125
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA…EGP B7RZ31_9GAMM/59-123
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA…KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA…NKP A0A0X3UC67_9GAMM/57-121
Showing Alignment Sequence Record
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIMVLAPRLTAKHPYDKVQDRNRK
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVADLMRKLDLDRPFKKLERKNRT
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVATVANQLRGRKRRAFARHREGP
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMAPMLIALNYRNRESHAQVDKKP
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMAPLFKVLSFRNREDQGLVNNKP
读取多个比对:通常,大多数序列比对文件包含单个比对数据,其中read()方法足以解析它。在多序列比对的情况下,比较两个以上的序列以获得它们之间的最佳序列匹配,并在单个文件中进行多序列比对的结果。如果序列比对格式有多个序列比对,则使用parse()方法而不是read()方法,它返回一个可迭代对象,可以迭代该对象以获取实际比对。下面给出了一个基本示例:
蟒蛇3
# Import libraries
from Bio import AlignIO
# Parsing Sequence Alignment
alignment = AlignIO.parse(open("PF18225_seed.txt"), "stockholm")
# Show alignment generator
print(alignment)
# Printing alignment
for alignment in alignments:
print(alignment)
输出:
SingleLetterAlphabet() alignment with 5 rows and 65 columns
AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM…NRK B3PFT7_CELJU/62-126
AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA…NRT K4KEM7_SIMAS/61-125
MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA…EGP B7RZ31_9GAMM/59-123
ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA…KKP A0A143HL37_9GAMM/57-121
TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA…NKP A0A0X3UC67_9GAMM/57-121