- BioPython 模块中的序列
- BioPython 模块中的序列(1)
- Biopython-安装(1)
- Biopython-安装
- Biopython-PDB模块
- Biopython-PDB模块(1)
- Biopython-序列比对
- Biopython-序列比对(1)
- Biopython – 序列比对
- Biopython-序列
- Biopython – 序列比对(1)
- Biopython-序列(1)
- Biopython教程
- Biopython教程(1)
- Biopython-简介
- Biopython简介
- Biopython简介(1)
- Biopython-简介(1)
- 讨论Biopython
- 讨论Biopython(1)
- Biopython – 成对对齐
- Biopython – 成对对齐(1)
- Biopython-绘图(1)
- Biopython-绘图
- Biopython - 序列操作
- Biopython - 序列操作(1)
- Biopython-序列I O操作(1)
- Biopython-序列I / O操作
- Biopython-测试技术(1)
📅 最后修改于: 2020-11-09 05:04:48 🧑 作者: Mango
BioSQL是一种通用数据库模式,主要用于存储所有RDBMS引擎的序列及其相关数据。它的设计方式可以保存来自所有流行的生物信息学数据库(如GenBank,Swissport等)的数据。它也可以用于存储内部数据。
BioSQL当前为以下数据库提供特定的架构-
- MySQL(biosqldb-mysql.sql)
- PostgreSQL(biosqldb-pg.sql)
- 甲骨文(biosqldb-ora / *。sql)
- SQLite(biosqldb-sqlite.sql)
它还提供对基于Java的HSQLDB和Derby数据库的最小支持。
BioPython提供了非常简单,轻松和高级的ORM功能,可与基于BioSQL的数据库一起使用。 BioPython提供了一个模块BioSQL来执行以下功能-
- 创建/删除BioSQL数据库
- 连接到BioSQL数据库
- 解析序列数据库,如GenBank,Swisport,BLAST结果,Entrez结果等,并将其直接加载到BioSQL数据库中
- 从BioSQL数据库中获取序列数据
- 从NCBI BLAST获取分类数据并将其存储在BioSQL数据库中
- 针对BioSQL数据库运行任何SQL查询
BioSQL数据库架构概述
在深入研究BioSQL之前,让我们了解BioSQL模式的基础。 BioSQL模式提供了25个以上的表来保存序列数据,序列特征,序列类别/本体和分类信息。一些重要的表格如下-
- 生物数据库
- 生物进入
- 生物序列
- 序列特征
- 分类单元
- taxon_name
- 反论
- 术语
- 外部参照
创建一个BioSQL数据库
在本节中,让我们使用BioSQL团队提供的模式创建一个示例BioSQL数据库,即biosql。我们将使用SQLite数据库,因为它非常容易上手并且没有复杂的设置。
在这里,我们将使用以下步骤创建一个基于SQLite的BioSQL数据库。
步骤1-下载SQLite数据库引擎并安装。
步骤2-从GitHub URL下载BioSQL项目。 https://github.com/biosql/biosql
步骤3-打开控制台并使用mkdir创建目录并输入。
cd /path/to/your/biopython/sample
mkdir sqlite-biosql
cd sqlite-biosql
步骤4-运行以下命令以创建新的SQLite数据库。
> sqlite3.exe mybiosql.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite>
步骤5-从BioSQL项目(/ sql / biosqldb-sqlite.sql`)复制biosqldb-sqlite.sql文件,并将其存储在当前目录中。
步骤6-运行以下命令以创建所有表。
sqlite> .read biosqldb-sqlite.sql
现在,所有表都在我们的新数据库中创建。
步骤7-运行以下命令以查看数据库中的所有新表。
sqlite> .headers on
sqlite> .mode column
sqlite> .separator ROW "\n"
sqlite> SELECT name FROM sqlite_master WHERE type = 'table';
biodatabase
taxon
taxon_name
ontology
term
term_synonym
term_dbxref
term_relationship
term_relationship_term
term_path
bioentry
bioentry_relationship
bioentry_path
biosequence
dbxref
dbxref_qualifier_value
bioentry_dbxref
reference
bioentry_reference
comment
bioentry_qualifier_value
seqfeature
seqfeature_relationship
seqfeature_path
seqfeature_qualifier_value
seqfeature_dbxref
location
location_qualifier_value
sqlite>
前三个命令是配置命令,用于配置SQLite以格式化方式显示结果。
步骤8-将由BioPython团队https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk提供的示例GenBank文件ls_orchid.gbk复制到当前目录,并将其另存为orchid.gbk 。
步骤9-使用以下代码创建一个Python脚本load_orchid.py并执行它。
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()
上面的代码解析文件中的记录,并将其转换为Python对象,然后将其插入BioSQL数据库。我们将在后面的部分中分析代码。
最后,我们创建了一个新的BioSQL数据库并将一些示例数据加载到其中。我们将在下一章中讨论重要的表格。
简单的ER图
生物数据库表位于层次结构的顶部,其主要目的是将一组序列数据组织到单个组/虚拟数据库中。生物数据库中的每个条目都指向一个单独的数据库,并且不会与另一个数据库混合。 BioSQL数据库中的所有相关表都引用了生物数据库条目。
bioentry表包含有关序列的所有详细信息,但序列数据除外。特定生物条目的序列数据将存储在生物序列表中。
taxon和taxon_name是分类法详细信息,每个条目都引用此表来指定其分类信息。
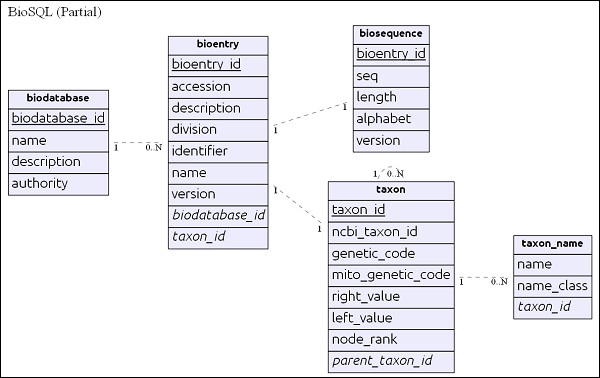
了解了架构之后,让我们在下一部分中研究一些查询。
BioSQL查询
让我们深入研究一些SQL查询,以更好地理解数据的组织方式以及表之间的相互关系。在继续之前,让我们使用以下命令打开数据库并设置一些格式化命令-
> sqlite3 orchid.db
SQLite version 3.25.2 2018-09-25 19:08:10
Enter ".help" for usage hints.
sqlite> .header on
sqlite> .mode columns
.header和.mode是用于更好地可视化数据的格式化选项。您也可以使用任何SQLite编辑器来运行查询。
列出系统中可用的虚拟序列数据库,如下所示-
select
*
from
biodatabase;
*** Result ***
sqlite> .width 15 15 15 15
sqlite> select * from biodatabase;
biodatabase_id name authority description
--------------- --------------- --------------- ---------------
1 orchid
sqlite>
在这里,我们只有一个数据库,兰花。
列出带有以下给定代码的兰花数据库中可用的条目(前3个)
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>
使用给定代码列出与条目(登录号-Z78530,名称-fasciculatum 5.8S rRNA基因以及ITS1和ITS2 DNA)相关的序列详细信息-
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>
获取与条目相关联的完整序列使用下面的代码(登录- C. fasciculatum 5.8S rRNA基因和ITS1和ITS2 DNA – Z78530,姓名) –
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>
列出与生物数据库,兰花相关的分类单元
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>
将数据加载到BioSQL数据库中
在本章中,让我们学习如何将序列数据加载到BioSQL数据库中。在上一节中,我们已经有了将数据加载到数据库中的代码,代码如下-
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()
我们将更深入地了解代码的每一行及其用途-
第1行-加载SeqIO模块。
第2行-加载BioSeqDatabase模块。该模块提供了与BioSQL数据库交互的所有功能。
第3行-加载os模块。
第5行-open_database使用配置的驱动程序(驱动程序)打开指定的数据库(db),并将句柄返回到BioSQL数据库(服务器)。 Biopython支持sqlite,mysql,postgresql和oracle数据库。
第6-10行-load_database_sql方法从外部文件加载sql并执行它。 commit方法提交事务。因为我们已经使用模式创建了数据库,所以可以跳过此步骤。
第12行-new_database方法创建新的虚拟数据库orchid,并返回一个句柄db以对orchi数据库执行命令。
第13行-加载方法将序列条目(可迭代的SeqRecord)加载到Orchid数据库中。 SqlIO.parse解析GenBank数据库,并将其中的所有序列作为可迭代的SeqRecord返回。加载方法的第二个参数(True)指示其从NCBI blast网站获取序列数据的分类法详细信息(如果系统中尚不可用)。
第14行-commit提交事务。
第15行-close关闭数据库连接并破坏服务器句柄。
获取序列数据
让我们从兰花数据库中获取一个标识符为2765658的序列,如下所示:
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))
在这里,server [“ orchid”]返回句柄以从虚拟数据库orchid获取数据。查找方法提供了一个根据条件选择序列的选项,我们选择了带有标识符2765658的序列。查找将序列信息作为SeqRecordobject返回。由于我们已经知道如何使用SeqRecord`,因此很容易从中获取数据。
删除数据库
删除数据库就像使用适当的数据库名称调用remove_database方法,然后按以下指定的方式提交一样简单-
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()